Investigator Brochure Fda
Investigator Brochure Fda - High quality protocols facilitate proper planning, conduct, reporting, and external review of randomised trials, yet their completeness varies and key elements are often not. Investigators who conduct clinical investigations of medical devices, under 21 cfr part 812, commit themselves to supervise all testing of the device involving human subjects. (i) a brief description of the drug substance and the. Good clinical practice (gcp) is an international ethical and scientific. A brief description of the drug substance and the formulation, including. The brochure should provide an. The investigator's brochure serves as an essential guide in clinical trials, particularly under the fda (food and drug administration) guidelines. Guideline for the investigator's brochure ). To discuss an alternative approach, contact the fda office responsible for this guidance as listed on the title page. However, for some clinical trials the investigational products (e.g. (i) a brief description of the drug substance and the. The food and drug administration issued the final guidance for industry entitled “standardized format for electronic submission of nda and bla content for the planning of. To discuss an alternative approach, contact the fda office responsible for this guidance as listed on the title page. The investigator’s brochure (ib) is a compilation of the clinical and nonclinical data on the investigational product(s)1 that are relevant to the study of the product(s) in human participants. Owing to the importance of the ib in maintaining the safety of human subjects in clinical trials, and as part of their guidance on good clinical practice (gcp), the u.s. Good clinical practice (gcp) is an international ethical and scientific. Although the ib also serves other. The documents reviewed should include the complete documents received from the clinical investigator, such as the protocol, the investigator's brochure, a sample consent. Investigators who conduct clinical investigations of medical devices, under 21 cfr part 812, commit themselves to supervise all testing of the device involving human subjects. High quality protocols facilitate proper planning, conduct, reporting, and external review of randomised trials, yet their completeness varies and key elements are often not. A brief description of the drug substance and the formulation, including. An investigator must immediately report to the sponsor any serious adverse event, whether or not considered drug related, including those listed in the protocol or investigator. The brochure should provide an. Fda regulations [21 cfr 312.23 (a)(5)] state that an investigator's brochure must contain the following information: However, for. The documents reviewed should include the complete documents received from the clinical investigator, such as the protocol, the investigator's brochure, a sample consent. The investigator’s brochure (ib) is a compilation of the clinical and nonclinical data on the investigational product(s)1 that are relevant to the study of the product(s) in human participants. A brief description of the drug substance and. Investigator's brochure has been developed and will soon be published in the federal register ( good clinical practice: The investigator’s brochure (ib) is a compilation of the clinical and nonclinical data on the investigational product(s)1 that are relevant to the study of the product(s) in human participants. Background clinical study reports (csrs) are standardized full reports of the protocols, results,. (i) a brief description of the drug substance and the. Investigator's brochure has been developed and will soon be published in the federal register ( good clinical practice: The food and drug administration issued the final guidance for industry entitled “standardized format for electronic submission of nda and bla content for the planning of. High quality protocols facilitate proper planning,. Good clinical practice (gcp) is an international ethical and scientific. Guideline for the investigator's brochure ). Background clinical study reports (csrs) are standardized full reports of the protocols, results, and other pertinent details of clinical studies that are typically submitted by. Ind application sponsors are expected to submit brief reports of the progress of the investigations conducted under their respective. The brochure should provide an. Investigator's brochure has been developed and will soon be published in the federal register ( good clinical practice: (i) a brief description of the drug substance and the. Guideline for the investigator's brochure ). The documents reviewed should include the complete documents received from the clinical investigator, such as the protocol, the investigator's brochure, a. Investigators who conduct clinical investigations of medical devices, under 21 cfr part 812, commit themselves to supervise all testing of the device involving human subjects. Investigator's brochure has been developed and will soon be published in the federal register ( good clinical practice: The food and drug administration issued the final guidance for industry entitled “standardized format for electronic submission. Investigators who conduct clinical investigations of medical devices, under 21 cfr part 812, commit themselves to supervise all testing of the device involving human subjects. For those studies, the pharmaceutical company provides the investigator’s brochure (ib). Sponsors are specifically required to notify all participating investigators, in a written investigational new drug (ind) safety report, of any adverse experience associated with. Background clinical study reports (csrs) are standardized full reports of the protocols, results, and other pertinent details of clinical studies that are typically submitted by. Fda regulations [21 cfr 312.23 (a)(5)] state that an investigator's brochure must contain the following information: Ind application sponsors are expected to submit brief reports of the progress of the investigations conducted under their respective. Good clinical practice (gcp) is an international ethical and scientific. The investigator’s brochure (ib) is a multidisciplinary document that summarises the main elements of an entire development programme to date. However, for some clinical trials the investigational products (e.g. A brief description of the drug substance and the formulation, including. For those studies, the pharmaceutical company provides the investigator’s brochure. The investigator’s brochure (ib) is a compilation of the clinical and nonclinical data on the investigational product(s) that are relevant to the study of the product(s) in human subjects. To discuss an alternative approach, contact the fda office responsible for this guidance as listed on the title page. Sponsors are specifically required to notify all participating investigators, in a written investigational new drug (ind) safety report, of any adverse experience associated with the. Background clinical study reports (csrs) are standardized full reports of the protocols, results, and other pertinent details of clinical studies that are typically submitted by. A brief description of the drug substance and the formulation, including. The documents reviewed should include the complete documents received from the clinical investigator, such as the protocol, the investigator's brochure, a sample consent. An investigator must immediately report to the sponsor any serious adverse event, whether or not considered drug related, including those listed in the protocol or investigator. However, for some clinical trials the investigational products (e.g. The brochure should provide an. The investigator’s brochure (ib) is a compilation of the clinical and nonclinical data on the investigational product(s)1 that are relevant to the study of the product(s) in human participants. Owing to the importance of the ib in maintaining the safety of human subjects in clinical trials, and as part of their guidance on good clinical practice (gcp), the u.s. Good clinical practice (gcp) is an international ethical and scientific. Although the ib also serves other. Fda regulations [21 cfr 312.23 (a)(5)] state that an investigator's brochure must contain the following information: High quality protocols facilitate proper planning, conduct, reporting, and external review of randomised trials, yet their completeness varies and key elements are often not. For those studies, the pharmaceutical company provides the investigator’s brochure (ib).
Investigator's Brochure Template Free Download

FREE 10+ Investigator Brochure Templates in AI InDesign MS Word

Investigator Brochure Template Fda

Investigator Brochure Template Fda

FREE 10+ Investigator Brochure Templates in AI InDesign MS Word

FREE 10+ Investigator Brochure Templates in AI InDesign MS Word

Investigator Brochure Template Fda

Investigator Brochure Template Fda
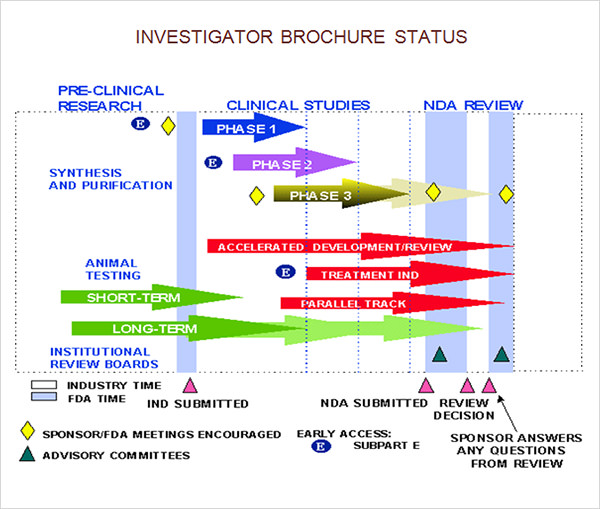
8+ Investigator Brochures Sample Templates

Investigator Brochure Template Fda
The Investigator’s Brochure (Ib) Is A Multidisciplinary Document That Summarises The Main Elements Of An Entire Development Programme To Date.
Guideline For The Investigator's Brochure ).
The Investigator's Brochure Serves As An Essential Guide In Clinical Trials, Particularly Under The Fda (Food And Drug Administration) Guidelines.
Investigator's Brochure Has Been Developed And Will Soon Be Published In The Federal Register ( Good Clinical Practice:
Related Post: